Le syndrome de Pfeiffer: causes, symptômes, traitement
Le syndrome de Pfayffer est extrêmement rareune maladie génétique qui survient en moyenne chez un nouveau-né sur 100 000. Affecte également souvent les garçons et les filles. Le symptôme principal de la maladie est la coalescence précoce des os du crâne dans le processus d'embryogenèse, à cause de laquelle le cerveau ne peut plus se développer normalement.
Informations générales
La maladie a été découverte par le célèbre Allemandle généticien Rudolf Pfeiffer. En 1964, il a décrit les symptômes d'une maladie jusqu'alors inconnue, dont les principaux signes étaient des anomalies de la structure du crâne et une fusion des doigts sur les pieds ou les mains (syndactylie). Le syndrome a été observé chez huit patients de la même famille, ce qui indiquait la nature génétique de la maladie. D'autres études ont montré que le syndrome de Pfeiffer est transmis par un type autosomique dominant.
L'échographie peut détecter cette maladie chez le fœtus dès le deuxième trimestre de la grossesse. À l'échographie, on observe des anomalies du développement du crâne, des organes internes et des extrémités.
Symptômes
En 1993, le généticien américain Michael CohenIl a proposé de classer le syndrome de Pfeiffer en trois types en fonction de la sévérité des symptômes. Maintenant, sa classification est généralement acceptée et largement utilisée dans la littérature médicale.
Les symptômes du syndrome de Pfayffer type 1 comprennentcraniosynostose - une condition dans laquelle une ou plusieurs sutures crâniennes fusionnent prématurément pour former un os intégral. Par conséquent, il y a des changements dans la forme du crâne et du visage - traits anormaux hypertelorism, oreilles basses. Il peut y avoir une détérioration de la vision, une augmentation de la pression intracrânienne, une fente palatine. Chez 50% des patients, la perte auditive est notée. La plupart des personnes atteintes de ce type de maladie ont des niveaux normaux d'intelligence et ne présentait aucune anomalie neurologique.
Chez les enfants atteints du syndrome de Pfayffer de type 1, externedes signes, tels que de larges doigts courts sur les bras et les jambes, sont observés presque toujours. La syndactylie est un symptôme possible mais non nécessaire de ce type de pathologie.
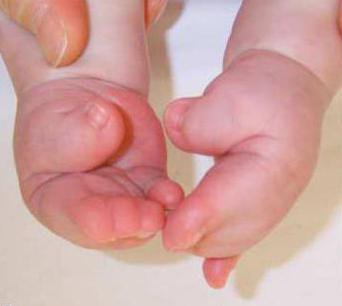
Ce type de syndrome est héréditaire autosomique dominant.
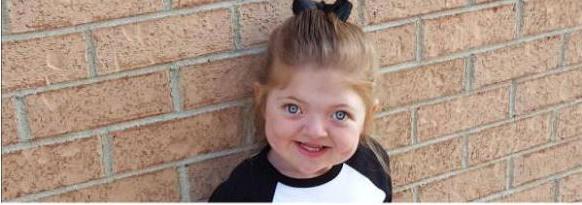
Voici une photo du syndrome de Pfeiffer. L'espérance de vie en Russie pour les personnes souffrant de cette maladie dépasse 60 ans.
Pour la maladie de type 2, le crâne se caractérise parforme de "trèfle", la fusion des vertèbres, la syndactylie et le déplacement des globes oculaires en avant (proptosis). Dès les premiers jours de la vie, des troubles neurologiques graves apparaissent.
Le troisième type de maladie est caractérisé encore pluscraniosténose précoce. Le crâne est anormalement allongé et ne forme pas de «trèfle». Il y a des anomalies des dents, de l'hypertélorisme, un retard mental, des malformations des organes internes.
Durée de vie au 2ème et 3ème typela maladie varie de plusieurs jours à plusieurs mois - les défauts des organes internes et les troubles neurologiques ne sont pas compatibles avec la vie. Les mutations responsables de ces deux types de syndrome de Pfeiffer sont sporadiques et ne sont pas génétiquement héréditaires.
Causes
Le syndrome de Pfayffer est associé à des mutations du récepteur 1le facteur de croissance des fibroblastes (FGFR1) sur le chromosome 8 ou récepteur du facteur de croissance des fibroblastes 2 (FGFR2) sur le chromosome 10. Les gènes qui influent sur la mutation responsable de la croissance normale des fibroblastes, qui joue un rôle clé dans le développement des os du corps.

Traitement
La médecine moderne ne permet pas encore d'éliminerdes mutations dans le génome humain, même si l'on sait dans quels gènes il y a eu perestroïka. Par conséquent, pour le syndrome de Pfeiffer, comme pour la plupart des autres maladies génétiques, le traitement est symptomatique. Pour les patients de type 2 et de type 3, le pronostic est défavorable: de nombreuses anomalies du développement ne peuvent être corrigées médicalement ou chirurgicalement.
Le syndrome de Pfayffer type 1 est compatible avec la vie. Le principal symptôme de la maladie - la fusion prématurée des os du crâne - peut être corrigé chirurgicalement. L'opération est recommandée pour les enfants jusqu'à l'âge de trois mois.
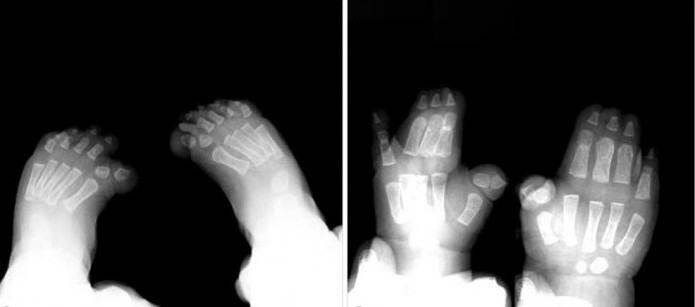
De plus, l'intervention chirurgicale est efficace dans le cas d'une syndactylie simple. Le traitement est préférable de commencer à l'âge de 18-24 mois, au cours de la croissance active des doigts et des orteils.